
本文转自“梅斯医学”公众号
CAR-T细胞疗法已经彻底改变了血液肿瘤的治疗格局,让许多原本无药可治的白血病、淋巴瘤患者获得了长期缓解甚至治愈。然而,仍有超过一半的患者会面临复发,其中最核心的难题之一就是CAR-T细胞的"耗竭"——它们在持续攻击肿瘤的过程中会逐渐失去战斗力,最终被肿瘤"拖垮"。
此外,部分CAR受体还会出现"无差别暴走",即使没有遇到肿瘤细胞也会持续发出本底信号(tonic signaling),导致T细胞提前衰老死亡。如何让CAR-T细胞像我们身体里的天然T细胞一样,既能精准杀敌,又能适时"收刀",避免过度消耗,一直是免疫治疗领域亟待解决的关键问题。
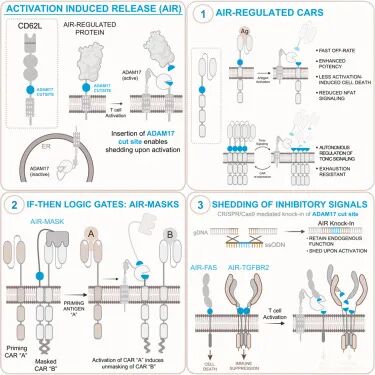
日前,一篇发表在国际顶级期刊Cell上的研究带来了突破性进展。来自斯坦福大学的科学家团队从人体T细胞自身的调节机制中获得灵感,发现了一个仅由15个氨基酸组成的"激活诱导释放"(AIR)基序。
将这个小小的"开关"插入CAR受体后,CAR-T细胞就能在被激活时自动被一种名为ADAM17的蛋白酶切割掉表面的受体,实现细胞自主的"踩刹车"。
这一设计不仅大幅减少了T细胞耗竭、延缓了激活诱导的细胞死亡(AICD),还在多种血液肿瘤和实体瘤模型中显著增强了抗肿瘤效果。更令人惊喜的是,这15个氨基酸的模块还能用来构建"双抗原确认"的逻辑门控回路,甚至精准改写内源性免疫抑制受体的调控方式,为下一代细胞免疫疗法提供了全新的设计范式。

从天然T细胞中偷师
天然T细胞拥有一套精妙的自我调节机制:当T细胞受体(TCR)被抗原激活后,会迅速下调表面受体的表达,避免过度激活导致的细胞死亡。研究团队决定模仿这一自然机制,首先系统地研究了T细胞激活后表面蛋白的变化规律。
他们使用环己酰亚胺(一种蛋白质合成抑制剂)处理人类原代T细胞,排除了新蛋白合成的干扰,专注于观察哪些蛋白会在激活后从细胞表面消失。
结果显示,T细胞激活后有数十种蛋白的表面表达显著下降,而这一过程主要由两种金属蛋白酶调控:ADAM10负责组成性的持续剪切,而ADAM17则只在T细胞被激活后才会发挥作用。由于ADAM17的"按需激活"特性完美契合了CAR-T细胞的调控需求,研究人员将目光聚焦在了寻找ADAM17特异性的剪切位点上。
他们筛选了40个已知的ADAM10/17底物,克隆了它们靠近细胞膜的25个氨基酸片段,插入到膜结合的mNeonGreen荧光报告蛋白中,在CD19.28ζ CAR-T细胞中进行测试。
最终,来自CD62L(一种T细胞表面的归巢受体)的15个氨基酸片段脱颖而出:它能在T细胞激活后被ADAM17高效且特异地剪切,完全不受ADAM10的影响。研究人员将这个模块命名为AIR(activation-induced release)。
实验显示,插入AIR模块的报告蛋白在T细胞激活后30分钟内,表面表达就下降了约80%;而当刺激停止后,仅需4-8小时,蛋白表达就能完全恢复到基线水平。即使经过多次"刺激-休息"循环,AIR模块依然能稳定工作,没有出现功能衰减。使用ADAM17特异性抑制剂可以完全阻断这种剪切,而ADAM10抑制剂则没有任何影响,进一步证明了AIR的高度特异性。
驯服“失控”的CAR
许多临床常用的CAR,尤其是高亲和力的GD2靶向CAR(HA.28ζ),存在严重的本底信号问题:即使没有遇到肿瘤细胞,CAR也会自发聚集并持续发出激活信号,就像一辆油门卡住的汽车,一直在空转耗油,很快就会导致T细胞耗竭和功能丧失。研究团队将AIR模块插入到HA.28ζ CAR的铰链区(位于跨膜区与抗原结合域scFv之间),结果令人振奋。
AIR-HA.28ζ CAR在T细胞表面的基础表达水平显著低于常规CAR,而且这种下调程度与CAR的本底信号强度成正比——本底信号越强,AIR介导的CAR下调越明显。
使用达沙替尼(一种Src激酶抑制剂,能阻断T细胞激活信号)处理后,AIR-HA.28ζ的表面表达迅速回升,证明这种下调完全依赖于CAR自身的本底信号激活。
更重要的是,AIR模块从根本上逆转了本底信号导致的T细胞耗竭。AIR-HA.28ζ CAR-T细胞表现出更低的激活标志物(CD25、CD69)和耗竭标志物(CD39、TIM-3、LAG-3、PD-1),同时富集了干细胞记忆T细胞标志物CD62L。
在体外实验中,它们增殖更旺盛、存活率更高、分泌的IL-2更多。在Nalm6-GD2白血病小鼠模型中,AIR-HA.28ζ CAR-T细胞的肿瘤清除能力显著优于常规CAR,小鼠的生存期也大幅延长。
转录组和质谱分析进一步揭示了AIR的作用机制:AIR-HA.28ζ CAR-T细胞的基因表达谱与经达沙替尼"药物休息"处理后的常规CAR-T细胞高度相似,两者共享了约78%的差异表达基因。
这意味着AIR模块实现了完全细胞自主的"间歇性休息",无需任何外源药物就能让T细胞保持在健康的功能状态。
即使没有本底信号的CAR,
AIR也能增强活力
对于不存在明显本底信号的CAR,比如临床上广泛使用的CD19.28ζ CAR,AIR模块同样能带来显著的性能提升。在静息状态下,AIR-CD19.28ζ的表面表达与常规CAR完全一致。但当遇到CD19⁺肿瘤细胞时,AIR模块会迅速介导CAR的剪切下调,避免了持续激活导致的激活诱导细胞死亡(AICD)。
在反复攻击肿瘤细胞的实验中,AIR-CD19.28ζ T细胞的凋亡率显著更低,扩增能力更强,肿瘤控制效果也更好。机制研究发现,AIR模块能选择性地调控CAR的信号输出:它保留了与细胞存活和增殖相关的NF-κB信号通路,同时适度降低了与耗竭和功能障碍密切相关的NFAT信号。使用ADAM17抑制剂可以恢复NFAT信号,进一步证明了这一机制。
研究人员还验证了一个关键问题:AIR介导的CAR剪切是否会影响对低抗原密度肿瘤的杀伤?结果显示,在低和中等抗原密度下,AIR-CAR的杀伤能力与常规CAR相当;而在高抗原密度下,AIR-CAR的清除效果反而显著更好。
此外,他们还证实,剪切下来的可溶性scFv片段不会结合并掩盖肿瘤细胞表面的抗原,也不会影响后续CAR-T细胞的杀伤功能,消除了这一潜在的安全隐患。
构建逻辑门控CAR
利用AIR模块快速、可逆的蛋白剪切特性,研究人员还设计出了精准的"与门"逻辑CAR,实现了"只有同时存在两种抗原时才会杀伤靶细胞"的功能,这将极大地降低CAR-T疗法的"靶向非肿瘤"毒性。
他们的设计思路是:将靶向EGFR的CAR与一个"掩蔽肽"共同表达,掩蔽肽通过高亲和力的亮氨酸拉链与CAR的scFv结合,物理阻挡其与EGFR的相互作用;而掩蔽肽本身也带有AIR模块。
同时,T细胞还表达一个靶向CD19的"触发CAR"。当只有EGFR⁺细胞存在时,掩蔽肽会一直阻断EGFR CAR的功能,T细胞不会发动攻击;只有当CD19⁺细胞同时存在时,CD19 CAR的激活会诱导ADAM17剪切掉掩蔽肽,暴露EGFR CAR的抗原结合位点,从而特异性杀伤EGFR⁺肿瘤细胞。
实验证明,这种AIR介导的逻辑门控响应速度非常快,仅需2小时的CD19刺激就能完全暴露EGFR CAR的结合位点。研究人员还验证了HER2和EGFR的抗原组合,证明这一策略具有良好的通用性。
改写内源性免疫抑制受体
AIR模块的应用远不止于调控CAR受体。研究团队进一步利用CRISPR-Cas9介导的同源定向修复技术,将AIR基序精确插入到内源性FAS(CD95)和TGF-β受体2(TGFBR2)的基因位点,实现了对这些免疫抑制受体的精准重编程。
改造后的T细胞在静息状态下保持正常的FAS或TGFBR2表达,维持正常的生理功能。但一旦被肿瘤抗原激活,这些受体就会被ADAM17快速剪切,从细胞表面消失。这样一来,T细胞在攻击肿瘤时就能避免FAS介导的凋亡和TGF-β介导的免疫抑制,而在静息状态下又不会出现自身免疫的风险。
实验结果令人惊叹:敲入AIR-FAS的CAR-T细胞在反复肿瘤挑战中的存活能力和抗肿瘤效果,与完全敲除FAS的细胞相当,但避免了FAS全敲可能导致的自身免疫性淋巴增殖综合征(ALPS)风险。
而AIR-TGFBR2 CAR-T细胞的表现更是超出预期:在存在高浓度TGF-β的肿瘤微环境中,它们的抗肿瘤效果甚至优于完全敲除TGFBR2的细胞。这是因为激活时脱落的TGFBR2可以像"分子海绵"一样结合并中和环境中的TGF-β,不仅保护了自身,还能改善整个肿瘤微环境的免疫抑制状态。
临床转化前景
这项研究的最大亮点在于,AIR模块仅由15个氨基酸组成,完全来源于人类自身的CD62L蛋白,免疫原性极低,几乎可以无缝整合到现有的任何CAR设计中,无需对CAR的其他部分进行大规模改造。
研究人员测试了多种临床常用的CAR,包括GD2.28ζ、GD2.BBζ、HER2.28ζ、HER2.BBζ等,无论它们本身的本底信号强弱,AIR模块都能显著提升它们的抗肿瘤效果,展现出了极强的通用性。
这一平台为下一代细胞免疫疗法带来了广阔的应用前景:首先,它能大幅提高CAR-T细胞的体内持久性,减少耗竭和激活诱导细胞死亡,从而降低患者的复发率。
其次,通过逻辑门控设计,它能实现更精准的肿瘤靶向,显著降低"脱靶"毒性。第三,AIR-TGFBR2等策略可以让CAR-T细胞抵抗肿瘤微环境中的免疫抑制,与PD-1/PD-L1抗体等免疫检查点抑制剂产生强大的协同效应。
第四,它能极大地简化CAR的开发流程,无需再针对每个scFv进行繁琐的本底信号优化,AIR可以为几乎所有CAR提供通用的"保驾护航"。最后,这一策略还可以拓展到CAR-NK、CAR-巨噬细胞以及TCR-T等其他免疫细胞疗法中。
当然,这项研究也存在一定的局限性。目前的实验主要基于免疫缺陷的NSG小鼠异种移植模型,尚需在免疫健全的动物模型和人体临床试验中进一步验证其安全性和有效性。
此外,AIR的剪切效率依赖于ADAM17的活性,在长期持续刺激下,ADAM17会被消耗,导致剪切效率下降;虽然AIR基序来源于人类蛋白,但插入到新的蛋白中可能会产生新的抗原表位,存在潜在的免疫原性风险。
但无论如何,这项研究为细胞免疫疗法的发展开辟了一条全新的道路。它告诉我们,与其从零开始构建复杂的合成电路,不如向大自然学习,挖掘和利用人体自身已经存在的精妙调控机制。这个仅15个氨基酸的"微小插件",或许将成为破解CAR-T细胞耗竭难题、推动免疫治疗进入新时代的关键钥匙。
参考资料:
[1]Jeremy R. Bjelajac et al, Cell-autonomous control of CAR signaling and receptor shedding via ADAM17-mediated proteolysis, Cell (2026). DOI: 10.1016/j.cell.2026.04.037.
